Despite rapid growth in research and development since their discovery at
Drexel University in 2011, the promising two-dimensional materials MXenes
face barriers to large-scale manufacturing and practical application due to
reliance on hazardous hydrofluoric acid etching methods. MXenes show
potential for uses including energy storage, catalysis, water purification,
optoelectronics, communication and healthcare, but scaling production while
controlling surface chemistry has challenged wide commercialization.
New research from scientists in the College of Engineering demonstrates a
pathway to sustainable, precise MXene synthesis using a computational
framework to guide fluorine-free production. The novel method, published in
Advanced Materials, highlights the power of computational
materials design to advance MXene manufacturing for real-world
technologies.
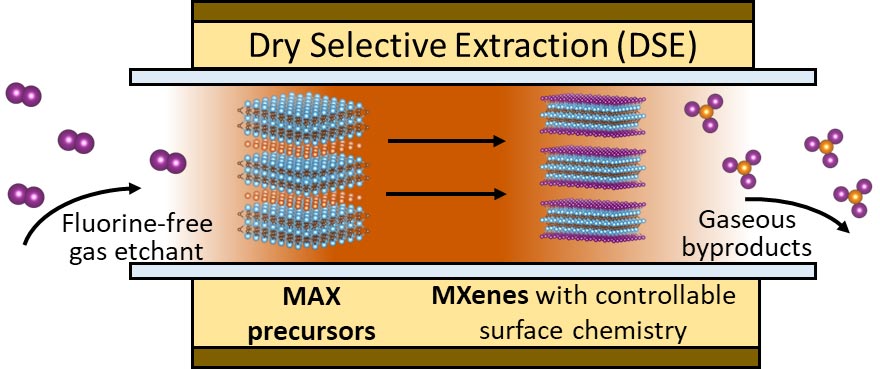
Schematic representation of this new synthesis pathway of MXene based on dry selective extraction (DSE).
Our motivation for using a computational approach is because it allows us
to design a new, more sustainable synthesis method efficiently and
rationally from a large space of materials chemistry and reaction
parameters,” explained
Yong-Jie Hu, PhD
, assistant professor of materials science and engineering at Drexel, who
led the computational study.
The study establishes a computational approach for dry selective extraction
of MXenes using iodine vapor to replace the hydrofluoric acid solutions.
The team used density functional theory (DFT) calculations to identify
thermodynamically stable candidates for the process, including the most
popular and widely used titanium carbide MXene.
“Identifying promising candidates of precursors through DFT-based
computational screening was challenging yet rewarding,” said Hu. “Our
calculations successfully revealed several promising candidates from
numerous possibilities.”
The CALPHAD (CALculation of PHAse Diagrams) approach was then used to model
optimal temperature and pressure conditions for iodine vapor etching
synthesis. Initial experiments produced the desired MXene composition,
validating the computational predictions.
“It is not enough to ‘synthesize’ a material in a computer. The process
must be transferred to a laboratory and, eventually, industrial-scale
production,” said co-author
Yury Gogotsi, PhD
, Distinguished University Professor and Charles T. and Ruth M. Bach
Professor at Drexel, who led the efforts of experimental validation.
This methodology opens the door to dry synthesis of MXenes with
compositions unattainable through conventional aqueous acid etching. It
also provides a framework to optimize synthesis conditions based on
thermodynamic characteristics. Moreover, the process is more sustainable,
as it does not require large amounts of water for synthesis and washing of
the product. The reaction byproducts can be collected and the iodine can be
reused in the synthesis process.
“We envision this methodology as a catalyst for advancing MXene synthesis
and applications,” said Gogotsi. “In the future, we plan to apply this
approach to other precursors and etching agents, expanding the range of
accessible MXene compositions and surface chemistries.”
The researchers see promise for improved performance and sustainability of
MXenes in various technological fields, driving innovation through the
computational design enabled by this study. “The ability to computationally
predict and control surface termination unlocks new possibilities for MXene
materials design,” concluded Gogotsi.
The multidisciplinary study was performed by a diverse research team,
including Eiara Fajardo, a fith year BS materials science and engineering
student; Ervin Rems, a master’s student from the European Erasmus Mundus
MESC+ program
; Mark Anaye, a PhD candidate studying under Gogotsi; Robert Lord, PhD, a
post-doctoral researcher in Gogotsi’s group; and David Bugallo, PhD, a
Marie Curie post-doctoral research in Hu’s group.
Read the full paper here: https://onlinelibrary.wiley.com/doi/abs/10.1002/adma.202305200
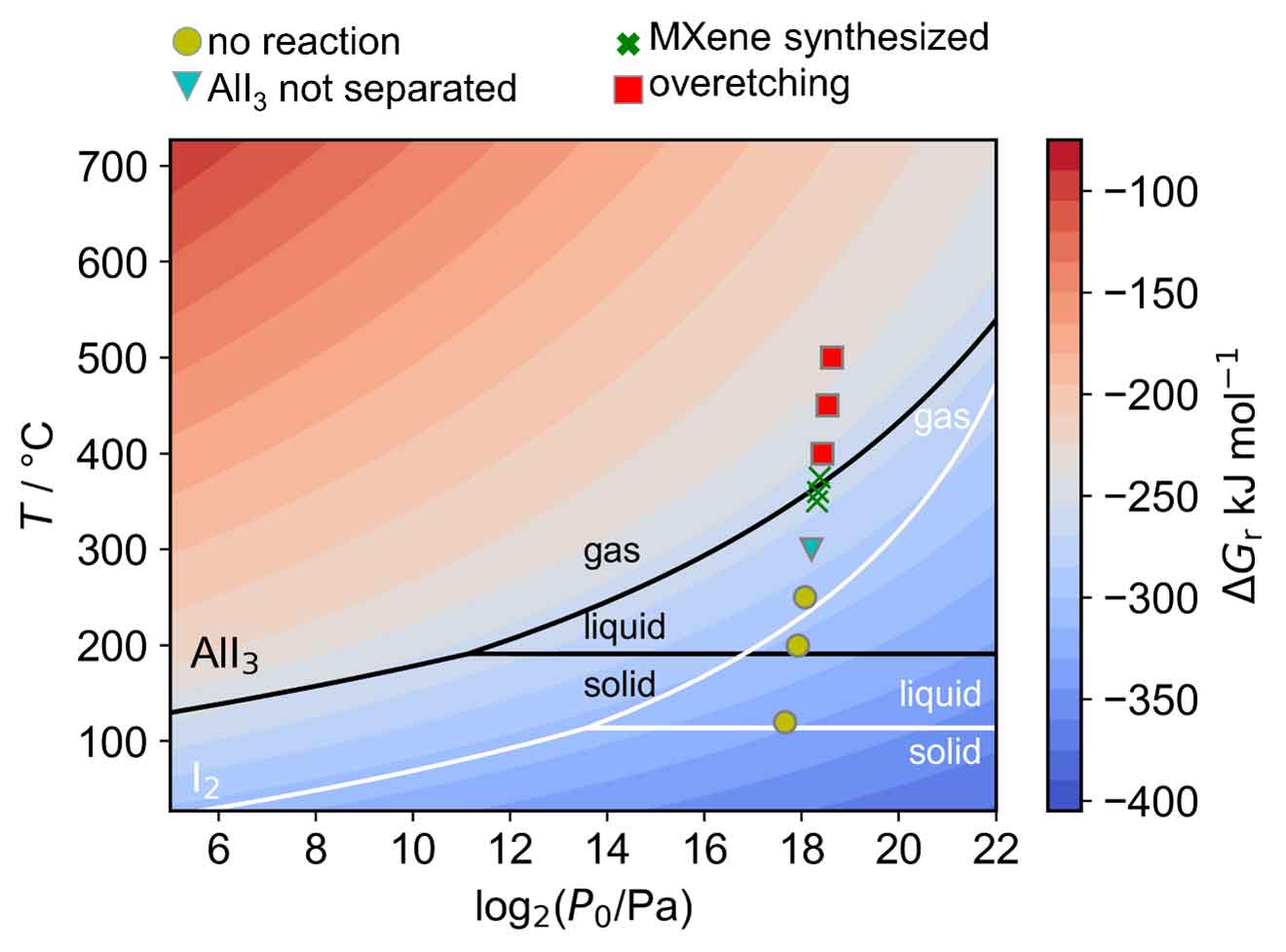 A computationally predicted reaction thermodynamic map to guide the discovery of optimal reaction temperature and pressure along with experimental validations.
A computationally predicted reaction thermodynamic map to guide the discovery of optimal reaction temperature and pressure along with experimental validations.