Research: Parkinson’s disease (PD) is a chronic and progressive neurological disorder affecting nearly 1 million people over the age of 60 in the U.S. and over 6 million people globally. Patients generally suffer from motor as well as a range of non-motor symptoms arising from the disease and treatment complications. Currently, there are no drugs on the market that can halt or reverse the progression of the disease. Levodopa is a gold standard for treating motor symptoms, but long-term usage leads to painful motor complications manifesting as Levodopa-induced dyskinesia (LID). Other treatment complications include development of tolerance, “ON-OFF” effects and worsening of several non-motor symptoms. Our laboratory is interested in understanding the neurobiological basis for LID, mild cognitive impairment (PD-MCI), and impulsive and compulsive behaviors associated with dopaminergic therapeutics.
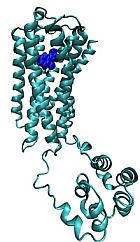
Biased signaling agonists of dopamine D3 receptor: Dopamine D3 receptors (D3R) are mainly localized to islands of calleja, olfactory tubercle, nucleus accumbens, and in the dorsal striatum. In the dorsal striatum, they are distributed on GABAergic medium spiny neurons and have a unique distribution that lends them to interact with D2Rs as autoreceptors and post synaptically with a subset of D1R-containing GABAergic neurons. Hence D3Rs form a component of both the direct and indirect pathways and play a significant role in the etiology of PD. D3R agonists can signal through G-proteins, β-arrestin, or both. We have recently designed and developed a novel series of G-protein-biased agonists that have high selectivity and specificity for D3Rs. These molecules were designed using a rational structure-based approach with our hybrid structure-based (HSB) method platform technology that utilizes biophysical modeling, simulations coupled with pharmacophore-based screening of drug-like small molecule libraries.
Biased signaling agonists to treat Levodopa-induced dyskinesia: The molecular mechanisms underlying the development of LID in PD are not well understood. LID is accompanied by a number of molecular changes including alterations in the levels and signaling of D1R, D2R and D3R receptors. In addition, we and others have hypothesized the role of D1R-D3R crosstalk in the dorsal striatum as one of the contributing factors to the etiology of LID. Our laboratory has recently demonstrated that SK609 a biased signaling agonist of D3R is effective in reducing the severity of LID when co-administered with Levodopa. We are currently testing the mechanism of action of SK609 in LID and its role in promoting D1R-D3R crosstalk in the dorsal striatum.
Biased signaling agonists to treat impulsive and compulsive behaviors: D3Rs are highly expressed in the corticostriatal pathways which include both limbic and the motor circuits that also mediate cognition, emotion, and reward processes in the brain. D3R agonism has been implicated to exacerbate compulsive and impulsive behaviors. It has been demonstrated that in addition to causing dyskinesias, chronic therapy with dopamine agonists can cause impulse control disorders (ICDs) in PD patients. ICDs comprise a group of complex behavioral disorders characterized by failure to resist an impulse or temptation to perform an act that is harmful to the individual or to others. Pramipexole (PRX), a known D3R agonist, has been shown to elicit ICDs in PD patients. Clinical manifestations of ICDs related to PRX therapy include pathological gambling, hypersexuality, compulsive shopping, and compulsive eating. While the etiology of ICD development in PD patients is not well understood, several clinical and pre-clinical studies have identified an association with D3R agonism. Our studies hypothesize that chronic treatment with D3R agonists that promote biased signaling via the β-arrestin pathway elicit ICDs. Our laboratory is currently testing this hypothesis using pharmacological, biochemical and behavioral assays in a hemiparkinson rodent model of PD. This study will provide insights into the molecular etiology of ICDs and the role of β-arrestin mediated pathways activated by D3R agonists which can be utilized to develop effective therapeutics for PD without the risk for ICDs.
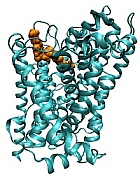
Screen for allosteric modulators of monoamine transporters: Monoamine transporters are an integral part of neurotransmitter homeostasis. Dysregulation of these transporters leads to neurological and psychiatric disorders. Most significant members of this family of transporters include Dopamine transporter (DAT) and serotonin transporter (SERT). Psychostimulants such as cocaine, methamphetamine and MDMA elicit their effect through either the inhibition (cocaine) or reversal (amphetamines) of transporter function DAT and SERT. In collaboration with Dr. Ole Mortensen, we have developed an alternative approach to block the effects of psychostimulants using allosteric modulation of these transporters. Using the HSB method, we have identified two compounds, KM822 and KM986, that bind to one of the allosteric sites, cause specific conformational changes, and modulate transporter interaction with psychostimulants and yet have no adverse effects on the normal transport of endogenous substrates. We are currently interested in understanding the effects of these allosteric modulators in blocking compulsive behaviors in PD and as tools to understand DAT-D3R interactions.
Catecholamine modulators to treat mild cognitive impairment: Parkinson’s disease is a neurodegenerative movement disorder that is also associated with a spectrum of non-motor symptoms. Mild cognitive impairment in PD (PD-MCI) consists of deficits in prefrontal-cortex (PFC) mediated tasks, which often manifest before the onset of motor impairment and negatively impact a patient’s quality of life. Specific cognitive processes that are frequently impaired in PD patients, such as sustained attention and cognitive flexibility, are regulated by catecholaminergic activity. Our lab has designed a novel compound SK609 – a dopamine D3R agonist and a selective norepinephrine transporter inhibitor. SK609 improves motor deficits in a rodent model of PD. We have continued to use SK609 to pharmacologically enhance poor cognitive function. We have observed an SK609-induced increase in sustained attention performance in both naïve low performing animals and PD rodents, suggesting that SK609 has the therapeutic potential to treat sustained attention deficits. This PD-MCI project also examines the role of D3R in cognitive flexibility under pathophysiological PD conditions. We are currently testing the effects of SK609 in a battery of cognitive tasks in a rodent model of PD.
Disease modifying therapies for Parkinson’s disease: Despite several advances in developing therapeutics for PD, there are no drugs to halt or reverse the progression of the disease. In an effort to reduce neurodegeneration, our laboratory has developed two distinct classes of small molecules that target two signaling pathways but both culminating in neuroprotection and blocking neurodegeneration. The orally available KA592 is designed to block inflammatory pathways that promote sustained inflammation and GT951 and its analogs are designed to reduce excitotoxicity in PD.
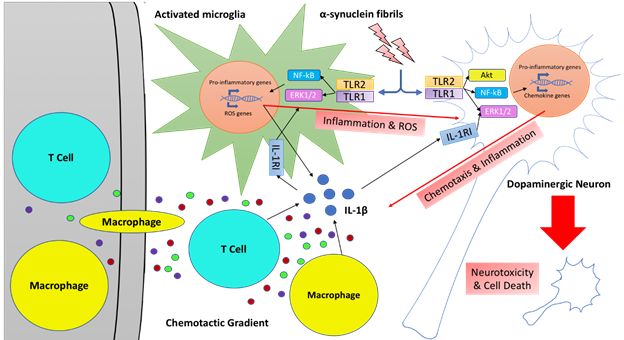
Neuroinflammation in PD: In addition to the loss of dopaminergic neurons in the substantia nigra pars compacta, PD pathology also includes the presence of Lewy Bodies (LB) and sustained inflammation in periphery and CNS. The presence of pro-inflammatory cytokines and activation of the microglia in the striatum are associated with neurodegeneration in PD. Fibrillar α-synuclein aggregates are known to selectively activate TLR2 receptors and drive neuroinflammation. In addition, maturation and activation of IL-1β can cause the production of pro-inflammatory cytokines in a MyD88 dependent signaling pathway. We hypothesize that pro-inflammatory signals produced by activation of IL-1β synergize with α-synuclein driven TLR2 signaling pathways to produce sustained levels of neuroinflammation that can exacerbate neurodegeneration in PD. To test this hypothesis, we are using a rodent model of PD induced by fibrillar aggregates of α-synuclein in combination with LPS treatment to test our newly designed IL-1R1 antagonist. We anticipate that our newly developed IL-1R1 antagonists can attenuate the sustained inflammation induced by both TLR2 and IL-1R1 and hence reduce neurodegeneration.
Excitotoxicity in PD: Loss of dopaminergic neurons in the substantia nigra pars compacta in PD leads to a cascade of events including an increase in extracellular glutamate levels resulting in hyperactivation of the glutamate receptors in the basal ganglia. Hyperactive glutamate receptors trigger activation of pathways that alter cell-cell interactions and produce toxic reactive oxygen species that affect the cell viability and neurodegeneration. We have recently designed a series of positive allosteric modulators of glutamate transporter – GLT1 (also called EAAT2) with high specificity to these transporters. We hypothesize that selective activation of GLT1 by our novel allosteric modulators can remove excess glutamate from the synapse thereby reducing excitotoxicity without inducing other glutamate receptor mediated toxic events. Blocking excitotoxicity has been shown to promote neuroprotection in PD. Our laboratory is currently evaluating these effects in a rodent model of PD.
Other interesting projects: Since the design of the HSB method a novel small molecule screening platform, we have designed small molecule modulators to a number of targets including HIV capsid, protein-protein interactions in Plasmodium falciparum and Toxoplasmosis gondii. Our pyrazole amide molecule 21A092 was licensed by medicines for malaria venture (MMV) and is currently in preclinical development.
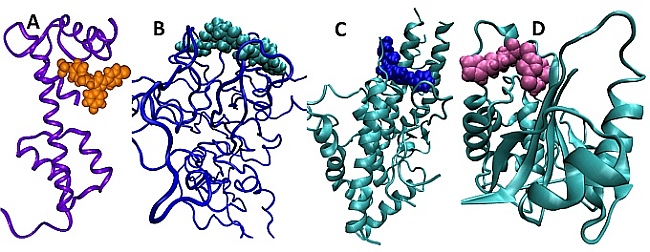
Design antimalarial compounds that target unique protein-protein interactions: Each year, there are an estimated 250 million cases of malaria worldwide, resulting in almost a million deaths, mostly in young children and pregnant women. Although several drugs are available for treating malaria, widespread resistance to most affordable drugs and the emerging resistance to alternative drugs, as well as their high cost, are major impediments in efforts to control malaria. Clearly, new affordable antimalarials with reduced propensity to develop resistance are needed urgently. The HSB method was applied to design small molecule inhibitors to A) MTIP-MyoA complex present in the inner membrane complex of Plasmodium falciparum B) apical membrane antigen-1 of P. falciparum that is involved in invasion of the parasite to red blood cells C) VAR2CSA protein – a member of the P. falciparum erythrocyte membrane protein-1 family responsible for pregnancy-associated malaria and D) the enzyme PfUCHL3 with dual activity as a deubiquitylase and a deneddylase.