Population genomics of bacterial pathogens:
Our work in comparative genomics focuses on how different strains of the same species of bacteria are phylogenetically related. How the population is structured based on the entire genomes can help inform researchers on how pathogenesis traits evolve. For example, in previous work we examined Moraxella catarrhalis, focusing genome sequences from 12 clinical isolates. The genome-based phylogenetic tree illustrated a ‘star-like’ pattern, indicating the population of M. catarrhalis was not separating into different clades, i.e. each genome was equally different from any other genome. However, all strains previously analyzed were resistant to killing in the presence of human serum. In our most recent publication we sequenced 22 new strains, 18 of which were sensitive to human serum. These new strains showed that M. catarrhalis can be broken into two distinct phylogenetic lineages correlating with their resistance to human serum. 4 strains previously thought to be M. catarrhalis were shown to differ by greater than 10% average nucleotide identity from the other M. catarrhalis strains, indicative of being a completely separate species.

Using the phylogenetic structure of these genomes, we used an algorithm called chromosome painting and were able to identify locations in the genome which showed evidence of recombination between lineages. These are regions which have a different pattern of phylogenetic relatedness, disagreeing with the overall phylogenetic pattern of the whole genome sequences.
The region of the genome with the highest recombination rate was at an amidotransferase subunit encoding gene, part of the translation apparatus. But remarkably, this gene was frequently flanked by an inserted beta-lactamase gene (providing antibiotic resistance), which had arrived by horizontal gene transfer.

Serum resistant and serum sensitive strains possessed this gene, and an examination of the phylogenetic tree based only on this region showed a very different lineage than observed with the whole genome data. This provided evidence that this gene, and the flanking regions were involved in HGT event(s) between and among the serum resistant and serum sensitive strains.
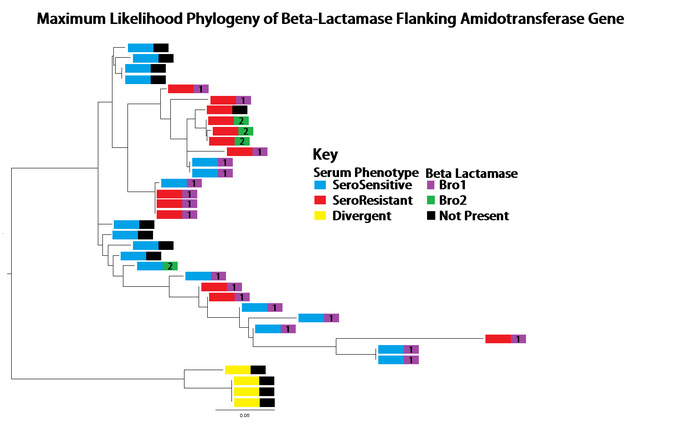
This is an example of the type of analyses our work in comparative genomics can find within bacterial communities. Future work in this area is focused on newly identified strains of M. catarrhalis. These show extraordinary virulence not previously seen in this pathogen. Our goal is to identify genomic characteristics that explain this phenotype. Organism projects part of our past work, and that we have data for future analysis include Gardnerella vaginalis, Haemophilus influenzae, Streptococcus pneumoniae, and Lactobacillus crispatus
Population analysis of microbial species:
My other area of research interest is examining the ecological structure of human microbial communities, or human microbiomes. Using Pacific Biosciences (PacBio) sequencing technology we are able to generate high quality long read data of bacterial 16s ribosomal subunit genes. The 16s gene has been become the standard for bacterial identification, since it is present in every bacteria and has alternating regions of high and low conservation. Using PacBio sequencing is novel in that most other sequencing technologies cannot produce the same read lengths, so therefore do not provide the same level of specificity that examining a full length gene can achieve. With this technology we have been able to perfectly separate and classify a mock community of 20 organisms. We are currently using this sequencing technology and analysis pipeline to examine such communities as samples from brain tissue, sino-nasal regions, cancer patients, and soil samples. I’m particularly interested in using these new high quality methods to characterize the diversity of human microbial communities and how this relates to bacterial pathogenesis.